

Abstract
Background:
Pearson Syndrome (PS) is an ultra-rare disease caused by de-novo mitochondrial DNA (mtDNA) deletions. Patients present at infancy with sideroblastic anemia and later develop a multisystem metabolic disorder, leading to death in early or late childhood. No disease-modifying treatments are available for PS. Ex-vivo enrichment of functional mitochondria into various cells has been previously demonstrated, as has inter-cellular mitochondrial transfer. In preclinical models of mitochondrial and lysosomal disorders, hematopoietic stem and progenitor cells (HSCs) have been shown capable of carrying and transferring normal organelles into diseased tissues, thereby altering disease phenotype. Here, we show enrichment of PS-derived HSCs with wild-type mitochondria, a process termed mitochondrial augmentation. We further report on three patients with PS treated with autologous HSCs following ex-vivo mitochondrial augmentation.
Methods:
Diagnosis of PS was confirmed by MLPA and deletion-specific dPCR. Colony formation assays were performed on PS patient-derived HSCs, prior to and after mitochondrial augmentation. HSC mobilization was performed with GCSF alone (n=1) or with plerixafor (n=2) prior to leukapheresis. Autologous CD34+ cells were positively-selected using a CliniMACS system, followed by ex-vivo mitochondrial augmentation of the cells with maternal cryopreserved mitochondria carrying normal mtDNA as confirmed by MLPA. Enriched cells were intravenously infused without conditioning. Level of heteroplasmy (relative normal to deleted mtDNA) was determined by deletion-specific dPCR of DNA from peripheral blood. Patients were followed for a period of up to 1 year including clinical and metabolic evaluations. Adverse events were reported as per CTCAE v4.03. Cellular mitochondrial function was studied on peripheral blood mononuclear cells (PBMCs) by ATP content, O2 consumption and flow cytometry for TMRE (tetramethylrhodamine ethyl ester) and MTG (mitotracker green).
Results:
Three patients were treated with production and safety data available, and in two patients efficacy data is available. PS-patient derived HSCs have a diminished capacity to form colonies in vitro (median, 360 colonies per 5x104 cells vs. 1090 in healthy donors). HSC colony forming capacity increased by an average of 30% after mitochondrial augmentation. Target cell dose (4x106 CD34+ cells/kg) was not reached despite two leukapheresis procedures in patients 1 and 2, who received 1.1 and 1.8 million CD34+ cells/kg recipient, respectively. Patient 3 received 2.8 million cells/kg following a single apheresis. Mitochondrial enrichment in the products was 156%, 162% and 114% for patients 1, 2 and 3. To date, the only treatment-related adverse events noted were leukapheresis related, including anemia, hypocalcemia and alkalosis.
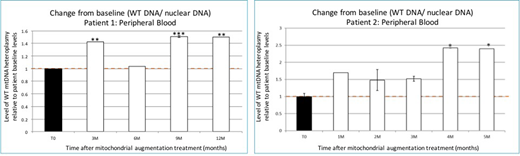
In two patients with more than 3 months follow-up, we observed in vivo mitochondrial enrichment starting 3-4 months after cellular therapy, and throughout the follow-up period (Figure). Metabolic function of PBMCs showed improvement at 5 months post-treatment in lymphocyte ATP content, O2 consumption and TMRE:MTG ratio, indicating improved mitochondrial respiratory capacity. Improvement in mitochondrial heteroplasmy and function was in line with clinical findings. Following cell therapy, no events of metabolic crisis occurred, along with normalization of a pre-treatment negative base excess in patient 1 and ongoing improvement in baseline lactate levels in patient 2. Aerobic ability and fine motor functions were superior compared to baseline in both patients. Importantly, quality of life, as measured by the International Pediatric Mitochondrial Disease Score (IPMDS), was greatly improved after treatment.
Conclusion:
We report a first in human study with a novel form of cellular therapy, mitochondrial augmentation, in which we enrich HSCs with organelles encoding non-mutated version of the mtDNA sequence. We show the ability of mitochondrial augmentation to improve in vitro PS-derived HSC function, and improvement in metabolic determinants, aerobic capacity and quality of life of two patients treated. Together, these preliminary clinical data suggest that mitochondrial augmentation therapy is safe, and may alter the clinical course for patients with mitochondrial deletions/mutations including PS.
Jacoby:Novartis Israel: Consultancy. Blumkin:Minovia Therapeutics: Employment. Sher:Minovia Therapeutics: Employment. Yivgi Ohana:Minovia Therapeutics: Employment. Toren:Novartis Israel: Consultancy.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal